
新闻活动
新闻动态
一文总结CGT 产品BLA 申报关注点 新药的研发过程历经早期研发、临床前研究、临床研发和上市及上市后持续追踪等过程,其中新药临床试验申请(Investigational New Drug, IND)和生物制品许可申请(Biologics License Application, BLA) 是2个关键的里程碑进展。药物通过临床前试验后,可向监管部门提交IND申请,以开展临床试验,包括药物系统性研究、不良反应及/或试验药物的吸收、分布、代谢和排泄等,确定试验药物在人体上的安全性与有效性。BLA申报需要递交生物制品的生产制造、非临床研究和临床研究等材料。 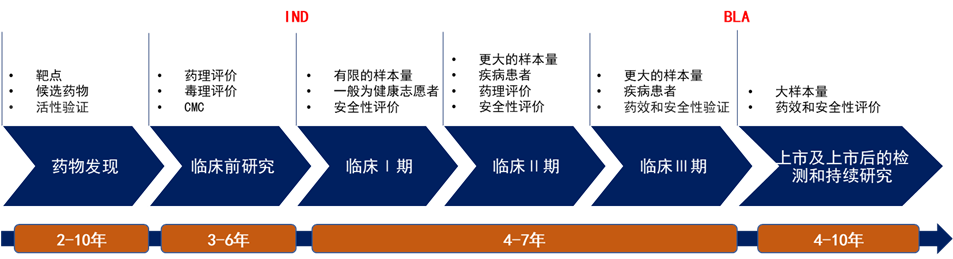 图1. 新药研发到上市流程。根据公开资料整理
细胞基因治疗(Cell and Gene Therapy, CGT)作为新一代突破性精准治疗手段,在生物医药领域尤其是癌症、遗传病及罕见病治疗领域极具发展前景。近年来随着CGT药物的快速推进,越来越多的药物进入了IND甚至是BLA阶段。据ASGCT 最新报告数据,全球临床试验增长显著。丁香园Insight 数据库也显示,从全球免疫细胞疗法进展迅速,2010年至2023年第三季度,全球共有2555 项免疫细胞疗法相关临床试验登记,其中中国1234项(48.3%),2016年起,中国已成为免疫细胞临床试验主要开展地区。中国细胞疗法发展迅速,2023年已有多个CAR-T、CAR-NK、TIL 、TCR-T获批IND(图3)。AAV 产品共有32款获NMPA批准临床,其中2023年就有16款,目前已有三款产品推进到临床Ⅲ期,分别是诺华的OAV101、信致医药的BBM-H901和纽福斯的NR082。随着中国CGT 药物的快速发展,未来将有多个CGT 药物进入BLA申请阶段。
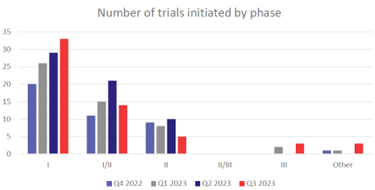 图2. 不同阶段启动的临床试验汇总。Source: Trialtrove| Citeline, October 2023  图3. 2023年IND获的细胞疗法汇总(部分)。数据来自CDE 官网和公开资料 那么CGT 药物BLA 申报前有哪些注意事项呢?根据对FDA 现场核查483缺陷项的统计,缺陷项出现较多的是:
1.质量系统缺陷,如偏差管理问题、纠正预防措施等;
2.设施设备问题,如环境监控不足、设备清洁和确认问题;
3.生产问题,如批记录不足、无菌工艺验证缺失或不充分等。
因此根据这几点,小编将从批生产、质量、设备、物料、数据完整性等多个维度为您总结BLA 申报前的关注点。
1. 工艺验证
FDA 在其指南中将工艺验证定义为:“从工艺设计(Process Design) 阶段到商业生产(Commercialization) 的数据收集和评估,建立科学证据表明工艺能够始终如一地提供优质产品”。工艺验证的目的就是确保药品始终符合质量标准,需覆盖杂质清除能力与检测能力、外源因子控制和工艺稳健性等多个维度。工艺验证的三个阶段分别是:
第1阶段工艺设计。包括工艺开发(如建立QTPP、初步识别CQA、定义生产工艺、初步稳定性研究)和工艺表征(包括工艺参数风险评估、建立缩小模型、工艺表征研究、杂质清除与最差条件研究等、原材料关键性评估与表征等,最终将形成工艺设计报告)。
第2阶段工艺确认:即评估工艺是否可重复,确保质量一致且可靠。包括中间体稳定性验证、溶液稳定性验证、填料清洁与保存、寿命验证、超滤膜包等清洁与寿命验证、PPQ运行、稳定性研究等。
第3阶段持续工艺验证:即在商业化生产中的持续验证,保证所有生产工艺都处于受控状态。在这个过程中,仍需不断收集数据,并进行SPC(Statistical Process Control) 分析,以便发现影响工艺稳定性的潜在因素。
质量管理
2. 无菌工艺模拟(APS)
由于CGT产品的特殊性,无法进行终端灭菌,尤其是细胞治疗产品和部分病毒产品甚至不能经过终端无菌过滤,因此需要全程无菌生产工艺,这就需要对整个生产过程进行无菌模拟,以证明确定的生产工艺能够持续且可重复的生产出符合要求的无菌产品。无菌工艺模拟试验需要在遵循相关法规标准的前提下,通过设计最差状况充分模拟生产工艺的全过程,即覆盖所有暴露操作,最大限度保障全流程无菌性。
2022 年 9 月,药明生基无锡基地在为客户上市产品的慢病毒载体生产进行了工艺转移和全程无菌工艺模拟试验,通过设计最差状况充分模拟了慢病毒载体生产工艺的全过程,确认了药明生基无锡基地慢病毒载体线的整体无菌保障水平,说明慢病毒载体产线的现有人员、洁净环境、设施设备、物料、操作规程符合全流程无菌生产的要求。
3. 生产设计方式
密闭或非密闭系统;
共线策略,共线生产管理包括从设备、厂房和公用设施、流程、监控、物料和人员等6个方面进行全面的风险评估,同时制定相应的管理要求。当有新产品引入时,共线生产会发起新的风险评估,并制定防交叉污染措施,确保新引入产品和原有产品之间没有共线生产的任何风险。
4. 无菌控制策略
需关注设施设备布局、人、物流;更衣管控;环境监控;消毒策略,消毒剂验证;操作控制;洁净区环境控制、污染与措施等多个方面。生产设施设备符合GMP法规要求,从使用、清洁消毒和维护等方面制定书面程序并严格执行,确保基础设施管理对产品的无菌控制无负面影响。生产相关人员加强培训,确保生产正常进行并确保污染和交叉污染得到有效控制。对生产用原材料进行严格的质量控制,降低CGT终产品中外源因子污染风险,保证产品安全性。
5. 排产方式
充分准备,制定合理的排产计划并有序实施和监控、关注最大产能、关注同时生产不同品种,不同批次,不同工序时如何制定排产计划。
药品生产应具备完善的质量体系,具有与药品生产相适应的组织机构,并能保证质量体系的有效运行。质量管理需关注以下几点:完整的组织机构、合理的培训机制、偏差处理、变更控制、纠正和预防措施(Corrective Actions and Preventive Actions, CAPA)有效性、年度质量回顾、应急处理程序等。
基于监管机构对CGT产品的法规要求,药明生基建立了一套完善的质量管理体系,包括质量体系、物料体系、设备和设施、包装和标签、生产体系和实验室体系等,6大系统相辅相成。整个质量管理体系的基本元素,包括质量管理评审、质量改进计划、纠正预防措施、变更管理、产品和工艺的系统绩效质量、质量风险管理和知识管理,来促进整个体系的持续改善。
质量控制
CGT 产品的质量控制主要集中在质粒与病毒载体、原辅料、生产和检定用细胞基质、菌毒种库等方面,包括中控、工艺参数与工艺性能监控、产品表征与放行、稳定性研究、物料控制、厂房设施与设备等。质量控制需关注以下几点:
1. 样品管理:取样管理、样品流管理、留样管理
2. 分析方法:验证与转移
3. 偏差管理:OOS、调查与CAPA
4. 产品质量属性:安全性、有效性、纯度杂质;关键质量属性的趋势分析
5. 稳定性研究:方案合理性,周期、代表性等;执行情况
我们将继续为大家分享在物料管理、厂房设施设备、数据可靠性、注册申报等几个维度上,我们需要重点关注哪些方面。
1.物料管理
在确证性临床阶段,物料管理应该关注以下方面:
1. 原辅料包材全检放行。根据相应EP/USP-NF/EP-USP/JP/Ch.P 等药典对原材料、辅料和内包材进行全检,如产品为无菌药品,其生产所用辅料和与产品直接接触的包装材料应当进行微生物(无菌)和细菌内毒素等安全性方面的检验。
2. 生产用原材料基于风险的检验,对用于基因和细胞产品生产用的主要物料开展入厂检验,并根据特定风险,考虑建立降低风险的措施,如加强质量控制等。
3. 物料稳定性研究。
4. 物料的包材相容性、包材密封性研究。
5. 供应商筛选、资料审核、现场审计、批准和年度审核。供应商质量体系符合性、注册批件、登记信息、物料质量证书或质量标准、必要的产品描述、物料的预期用途等资料审核。
6. 供应商分类评级。
2.厂房设施设备
厂房设施设计的核心问题是防止污染和交叉污染、防止混淆、防止差错。应该关注以下方面:
1. 厂房布局:采用隔离系统或密闭系统,并定期检查密闭系统或设备的完整性;洁净级别与布局;直接用于细胞产品生产的基因修饰病毒载体应与细胞产品、其他载体或生物材料相隔离,分别在各自独立的生产区域进行生产,并配备独立的空调净化系统。
2. 设备:关键设备的验证情况和维护状态,如细胞分离/分选、培养箱、生物反应器等。细胞产品生产用的密闭系统或设备,应定期检查其完整性。
3. 共线:共线评估;清洁程序。共线生产需要进行风险评估并制订防交叉污染措施。
4. 公用系统:校验与验证、运行与维护。
5. 无菌工艺区的环境监测系统
3.数据可靠性
1. 原始记录、图谱和数据的完整性与可靠性,可追溯且与申报资料保持一致。
2. 制定详细的控制标准,严格管控数据操作规程和人员授权。
3. 电子数据格式与仪器设备的匹配性。
4. 检验仪器计算机化系统用户分级权限管理,开启审计追踪功能,数据定时备份,防止数据修改、删除、覆盖等。
5. 产品的标识性和可追溯。采用经验证的计算机化系统,建立产品标识和追溯系统,确保在供者材料运输、接收以及产品生产和使用全过程中,来源于不同供者的产品不会发生混淆、差错,确保供者材料或细胞与患者之间的匹配性,且可以追溯。
4.注册申报
CGT产品的药品上市许可申请,需要准备支持药品上市注册的药学、药理毒理学和药物临床试验相关研究资料,确定质量标准,完成商业规模生产工艺验证,并做好接受药品注册核查检验的准备。提交的申报材料不仅要满足技术审评需要,还要满足核查机构关于注册核查资料的相关要求,因此原则上可以向药审中心提出沟通交流申请,探讨现有的研究数据是否满足药品上市许可的技术要求。同时,应正确评估相关规格申报的合理性、申报资料的准确性,注意按时提交补充材料等。
为加快药品的上市注册,药审中心设置了快速通道,以中国和美国为例说明:
1. 国家药品监督管理局药品审评中心(CENTER FOR DRUG EVALUATION, NMPA):对符合条件的药品注册申请,可以申请适用突破性治疗药物、附条件批准、优先审评审批及特别审批程序。
突破性治疗药物审评工作程序一般是在临床Ⅰ/Ⅱ期间提出,CDE接到申请后45个工作日(2个月左右)内反馈意见,公示无异议后CDE对纳入突破性治疗药物程序的药物优先配置资源进行沟通交流,加强指导并促进药物研发;
附条件批准程序, 申请人可以在药物临床试验期间,经充分评估后,可以提出早期沟通交流申请, 在提交上市许可申请前再次提出沟通交流(一般为二类会议,如已获得突破性治疗认证的品种直接是一类会议)。附条件批准申请应提交上市后临床研究计划、明确研究完成日期、最终临床研究报告提交日期,并承诺按时完成所有的临床试验。附条件批准所附的上市后研究应证明药品在适应症中的临床获益。
优先审评审批程序,经沟通交流(pre-BLA) 确认后,申请人应当在提出药品上市许可申请的同时提交,CDE 五个工作日内反馈,临床急需境外已上市境内未上市的罕见病药品,将直接纳入优先审评审批程序,不再对外公示,CDE 对纳入优先审评审批程序的上市许可申请,按注册申请受理时间顺序优先配置资源进行审评。对纳入优先审评审批程序的药品上市许可申请,审评时限为130日,其中临床急需的境外已上市境内未上市的罕见病药品审评时限为70日。
2. FDA生物制品评估和研究中心(CBER,Center for Biologics Evaluation and Research):对符合条件的药品注册申请,可使用快速通道指定(fast track designation)、突破性疗法指定(breakthrough therapy designation)、加速批准指定(accelerated approval)和优先审评指定(priority review designation)等快速途径来获得审批。
快速通道指定(fast track designation)程序旨在促进药物的开发和加快药物审批,以治疗严重疾病和填补未满足的医疗需求。申请人可以在药物开发过程中的任何时间发起,FDA受理审查该申请,并在60天内给出反馈意见。
突破性疗法指定(breakthrough therapy designation)旨在治疗严重疾病,且初步临床证据表明该药物可能在临床重要终点比现有疗法有显著优势。原则上,申请人应该在不迟于2阶段结束会议之前向FDA 提交申请,FDA将在收到申请后60天内作出答复。
加速批准指定(accelerated approval):FDA于1992年制定了加速批准法规,允许使用替代终点批准用于治疗严重疾病或满足了未满足临床需求的药物。
优先审评指定(priority review designation):FDA 的审批途径有优先审评或标准审评两种,相比NDA的标准审批需要10个月,优先审批会在6个月内进行审批。申请人可以按照《Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics》要求优先审查,FDA在收到原始BLA、NDA或疗效补充材料后60天内反馈。
关于药明生基
药明生基是药明康德旗下专注于细胞和基因疗法的CTDMO (Contract Testing, Development and Manufacturing Organization),致力于加速和变革细胞和基因疗法及其他高端疗法的研究、开发、测试、生产和商业化。药明生基能够助力全球客户将更多创新疗法早日推向市场,造福病患。更多信息,请访问www.wuxiatu.com
|

TOP
电话:+86(510)8858 3666
邮箱:info_atu@wuxiapptec.com
地址:江苏省无锡惠山经济开发区惠山大道1719号惠山生命科学产业园D7-5层




